Die Bedeutung der Biotechnologie für unser Leben nimmt permanent zu. Gleichzeitig interessieren sich immer mehr Investoren für Investments in Biotech-Startups. Dabei spielt die Unternehmensbewertung eine entscheidende Rolle. Welche Besonderheiten es bei biotechnologischen Unternehmen gibt, erläutert Dr. Marcus Furch von Rodos Biotarget in einem Gastbeitrag.
Immer mehr Startups übernehmen bei der Forschung und Entwicklung neuer biotechnologischer Anwendungen eine bedeutende Rolle, wie zahlreiche Partnerschaften und Lizensierungen mit großen Pharma-Konzernen zeigen. Ebenso interessieren sich institutionelle Investoren und VC-Gesellschaften wie der High-Tech Gründerfonds (HTGF) zunehmend für Investments in Biotech-Unternehmen.
Auch bei Seedmatch präsentieren sich immer wieder Biotech-Startups der Crowd: Riboxx, Oaklabs und OvulaRing konnten die Crowd bereits von sich überzeugen. Nun steht mit Rodos Biotarget ein weiteres Unternehmen der Branche in den Startlöchern für ein Crowdfunding. Einen ersten Eindruck vom Startup erhalten Sie hier. Dr. Marcus Furch, CEO und Co-Gründer von Rodos Biotarget, weist in seinem Gastbeitrag auf die Besonderheiten bei Investments in und der Bewertung von Biotech-Unternehmen hin.
Motivation und Expertise
 Es ist mir ein großes Anliegen, bei privaten Investoren ein besseres Verständnis über die Wertentwicklung eines pharmazeutischen Produktes wie einem Medikament, Impfstoff oder eines Diagnostikverfahrens zu erreichen, um gleichzeitig auch die Chancen, die ein Investment in Biotech-Unternehmen birgt, aufzuzeigen. Zu diesem Thema habe ich daher auch schon bei Business-Angel-Netzwerken vorgetragen.
Es ist mir ein großes Anliegen, bei privaten Investoren ein besseres Verständnis über die Wertentwicklung eines pharmazeutischen Produktes wie einem Medikament, Impfstoff oder eines Diagnostikverfahrens zu erreichen, um gleichzeitig auch die Chancen, die ein Investment in Biotech-Unternehmen birgt, aufzuzeigen. Zu diesem Thema habe ich daher auch schon bei Business-Angel-Netzwerken vorgetragen.
In meiner Funktion als Head of Multiproject Controlling in einem Medtech- und Pharmakonzern ab 2002 verantwortete ich die kaufmännische Planung und Steuerung unserer Therapieentwicklungsprojekte sowie die Bewertung einzelner Projekte und des gesamten Portfolios.
Zusammen mit einer beauftragten Unternehmensberatung führte ich mit der rNPV-Methode (risk-adjusted net present value) einen Bewertungsansatz ein, der sich in jener Zeit im Investment Banking und in vielen Pharmaunternehmen etablierte, weil sich damit auch frühe Entwicklungsprojekte einer Pipeline möglichst risikogerecht bewerten lassen.
Wie Biotech-Unternehmen bewertet werden
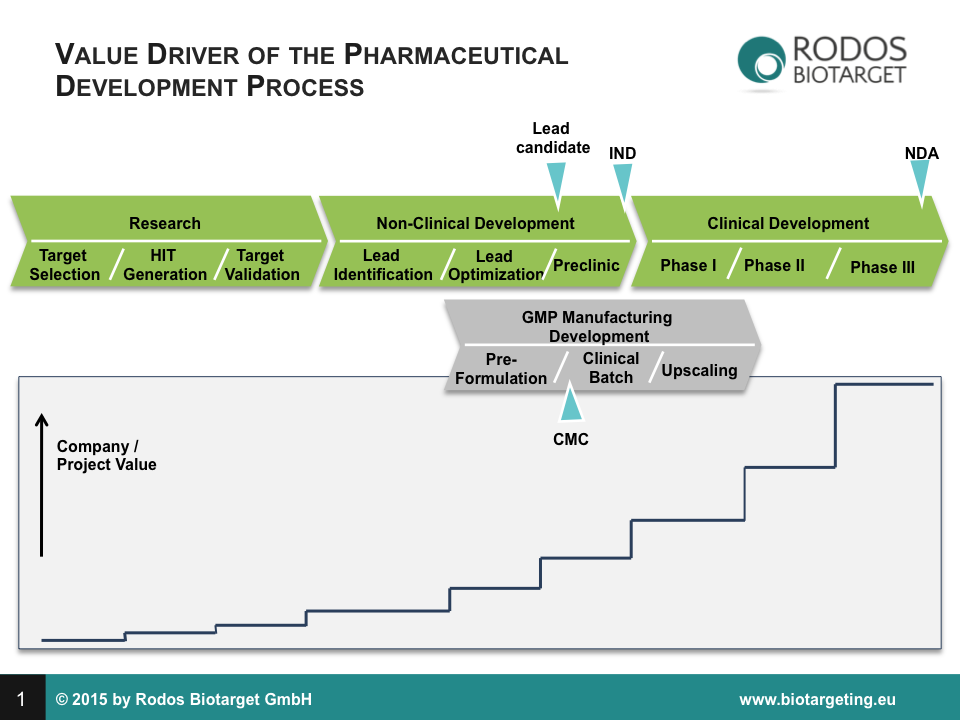
Der Wert eines pharmazeutischen Produktes oder eines biopharmazeutischen Unternehmens, welches sich als die Summe solcher Produkte zusammensetzt, entwickelt sich in diskreten Schritten. Die Schritte entsprechen dem Abschluss von wohl definierten Perioden, im Groben: Forschungsphase, Präklinische Entwicklung, Klinische Phase I, Klinische Phase II, Klinische Phase III (siehe Abb. unten).
Mit jedem der beschriebenen Entwicklungsschritte, die durch diskrete Meilensteine (wie z. B. anhand behördlicher Genehmigungen) abgeschlossen sind, steigt die Wahrscheinlichkeit, dass das pharmazeutische Produkt den Markt erreichen wird. Nach Abschluss der Präklinik liegt die mittlere Wahrscheinlichkeit des Markteintritts bei 16 bis 30 %, nach Abschluss von Phase I bei 30 bis 45 %, nach Abschluss von Phase II bei 45 bis 65 %, nach Abschluss von Phase III und vor der Zulassungsphase bei 65 bis 85 %.
Mit jedem erreichten Meilenstein sinkt somit das Risiko der Nichtzulassung, welches z. B. in einer risikoadjustierten Bewertung der Entwicklung nach der Nettobarwertmethode (rNPV-Methode) zugrunde gelegt werden kann. Damit steigt der inkrementelle Wert der Entwicklung und des entwickelnden Unternehmens. Einen erweiterte Einführung in die rNPV-Methode liefert ein Beitrag des Fachmagazins nature biotechnology.
Allerdings gibt es auch Risikounterschiede zwischen Entwicklungen in derselben Entwicklungsphase (daher sind zuvor auch Wahrscheinlichkeitsspannen angegeben), die u. a. abhängig sind von der Komplexität eines Ansatzes, der Wirkstoffklasse, dem postulierten Wirkmechanismus, dem Indikationsgebiet, der Entwicklungsdauer und der Entwicklungskosten über den gesamten Prozess.
Wird mit der Entwicklung eine Verbesserung eines bereits zugelassenen Medikaments angestrebt, z. B. dadurch, dass es in einer neuen Darreichungsform gegeben wird (Neuformulierung), wird diese prinzipiell als weniger riskant eingeschätzt, als für eine Neuentwicklung von Beginn mit innovativen Wirkstoffen aus einer wenig getesteten Wirkstoffklasse und / oder für die ein neuartiger Wirkmechanismus postuliert werden muss.
Wissenschaftlicher Exkurs: Der pharmazeutische Entwicklungsprozess am Beispiel eines innovativen Medikamentes
Die pharmazeutisch-industrielle Produktentwicklung ist streng reguliert. Der Prozess ist in wohldefinierte Periodenabschnitte und Arbeitspakete unterteilt; er erstreckt sich über mehrere Jahre. Zu Beginn steht die Forschungs- und Entwicklungsperiode: in ihr wird der Interventionspunkt eines Wirkstoffs (target) ausgewählt. Als Interventionspunkte eignen sich alle Moleküle, die auf einem Stoffwechselweg liegen, der pathologisch verändert ist. Als Target-Moleküle eignen sich u. a. extrazelluläre Botenstoffe, Zellrezeptoren, intrazelluläre Signaltransduktionsproteine sowie auch Gensequenzen.
Unser zunehmend besseres Verständnis um die molekularen Strukturen dieser Targets und ihrer Interaktion ermöglicht ein immer effizienter und schneller abstimmbares Design von Wirkstoffkandidaten. Viele als hits bezeichnete Wirkstoffkandidaten werden in dieser Phase parallel in schnellen In-vitro-Verfahren auf ihren Einfluss auf den ausgewählten Interventionspunkt getestet. Während die meisten hits wieder verworfen werden, kann das gewählte Target so als Angriffspunkt eines möglichen Wirkstoffes validiert werden. Dieser Abschnitt der Forschung und Entwicklung dauert bis zu acht Jahren und ist von vielen Versuchen und Irrtümern geprägt; darum liegt hier auch das größte Risikopotenzial.
Vielversprechende Wirkstoffkandidaten (leads) treten in die nächste Periode der nicht-klinischen Entwicklung ein und werden nun gezielt hinsichtlich ihres Wirksamkeits- und Toxizitätsprofils sowie ihrer Verfügbarkeit und Darreichungsform optimiert. In diesem Entwicklungsabschnitt kommen krankheitsrelevante Modelsysteme – z. B. komplexe Zell-Assays, in-vivo- und ex-vivo-Tests – zum Einsatz. Der beste Wirkstoffkandidat (lead candidate) wird in die Phase der späten oder eigentlichen Präklinik eingeschleust. Zu den entscheidenden präklinischen Testungen zählen bestätigende mehrmonatige Sicherheits-Toxizitäts-Studien in zwei Tierspezies (Nager und Nicht-Nager) sowie ausführliche Verteilungsstudien.
Idealerweise wird nach Abschluss der Präklinik auf der Grundlage der eingereichten erforderlichen regulatorischen Dokumente [u. a. das sog. Investigational Medicinal Product Dossier (IMPD) in Europa oder Investigational New Drug (IND) Application in den USA] an die in Deutschland zuständige Zulassungsbehörde (entweder das BfArM oder das Paul-Ehrlich-Institut) die Genehmigung für eine klinische Phase-I-Studie erteilt. Jedoch muss das Studienprotokoll des beantragenden Unternehmens zuvor behördlich genehmigt werden [die sog. Clinical Trial Authorization (CTA)].
Es schließt sich nun die Periode der klinischen Prüfung am Menschen an. In einer Phase-I-Studie werden die zuvor im Tier durchgeführten Experimente zur Sicherheit und Toxizität nun erstmals an gesunden menschlichen Probanden bestätigt. Zudem wird untersucht, wie ein Wirkstoff aufgenommen, verstoffwechselt (metabolisiert) und wieder ausgeschieden wird (Pharmakokinetik) und wie er dabei auf den Körper wirkt (Pharmakodynamik). In der anschließenden Phase II werden diese Experimente im Wesentlichen in Patienten wiederholt und dabei auch erste Erkenntnisse über die therapeutische Wirksamkeit des Medikaments gesammelt.
Die Wirksamkeit wird bei gleichzeitiger akzeptabler Verträglichkeit schließlich in größeren Patientenkohorten mit statistisch relevanten Fallzahlen in einer Phase-III-Studie mit den am Markt befindlichen therapeutischen Goldstandards verglichen. Nach positiver Bewertung durch die zuständige regulatorische Behörde kann der pharmazeutische Hersteller die Zulassung des Medikaments beantragen [New Drug Application (NDA)].
Zusätzlich zur Entwicklung und Validierung eines Wirkstoffs muss der pharmazeutische Hersteller rechtzeitig die industrielle Herstellung und Verpackung des Medikamentes nach geltenden Standards und Richtlinien (cGMP = current Good Manufacturing Practice) sicherstellen. Hiernach muss ein pharmazeutisches Produkt in einem dokumentierbaren kontrollierten und skalierbaren Herstellungsprozess erzeugt werden. Die erfolgreiche Überführung von der laborskaligen Produktion in einen cGMP-Prozess wird gegenüber den Behörden durch ein Chemicals, Manufacturing and Controls (CMC)-Dossier belegt.
Fazit
Mit dem Meistern der einzelnen Entwicklungsphasen nimmt der Wert eines Wirkstoffes zu, weil sich das Ausfallrisiko minimiert und entsprechend die Chance erhöht, dass das Medikament nach Zulassung in den Markt eingeführt werden wird. Der Wert eines pharmazeutischen Produktes oder eines biopharmazeutischen Unternehmens mit solchen Produkten nimmt stufenweise zu. Mit den größten Stufen der Wertsteigerung kann man in der Periode der klinischen Prüfung rechnen. Die Rodos Biotarget GmbH steht kurz vor Beginn der klinischen Phase-I-Studie und damit vor genau diesen Stufen. Mit der Unterstützung der Crowd soll die klinische Studie nun begonnen werden.
Warnhinweis: Der Erwerb dieser Vermögensanlage ist mit erheblichen Risiken verbunden und kann zum vollständigen Verlust des eingesetzten Vermögens führen.


